Quelle n’est pas la surprise du voyageur en Namibie qui se retrouve dans un décor surprenant, à mi-chemin entre les cratères lunaires et la savane africaine. Les cercles de fées s’étendent sur des milliers de kilomètres carrés de terres arides de Namibie. Ce sont des cercles de 2 à 10 mètres de diamètre, dont la délimitation est souvent constituée de touffes d’herbes pérennes du genre Stipagrostis et dont le centre est dépourvu de végétation. Ces cercles sont disposés régulièrement, tous les 5 à 10 mètres. Ces structures semblent stables et pourraient être établies depuis des centaines d’années.

Cercles de fées en Namibie (Crédit : Stephan Getzin, commons.wikimedia.org, CC-BY-SA 3.0)
Les cercles de fées intriguent depuis longtemps et posent de nombreuses questions. Comment se forment-ils et se maintiennent-ils au niveau individuel ? Comment forment-ils des structures si régulières au niveau du paysage ?
En premier lieu, l’hypothèse de colonies de termites qui seraient localisées au centre des cercles a été écartée. Des travaux sur des structures similaires dans le désert du Néguev, en Israël, suggèrent une autre hypothèse : chaque cercle de fées y est formé d’une unique plante du genre Stipagrostis qui émettrait des rhizomes souterrains. L’épuisement des ressources entrainerait la disparition des pousses aériennes au centre tandis que de nouvelles pousses se développeraient à la périphérie. Une troisième hypothèse incrimine l’avantage des plantes à la périphérie par rapport aux plantes au centre. Des modèles mathématiques montrent l’importance du rapport entre les flux d’eau et la dispersion de la biomasse. Cette dernière hypothèse semble aussi plus plausible en cas de clonalité que de cercles de fées constitués d’individus génétiquement différents.
Le collectif de chercheurs d’Afrique du Sud, d’Allemagne et des Etats-Unis de Christian Kappel et ses collaborateurs a testé l’hypothèse de clonalité des plantes constituant un cercle de fées. Leurs travaux ont été publiés dans le journal Communications Biology en novembre 2020.
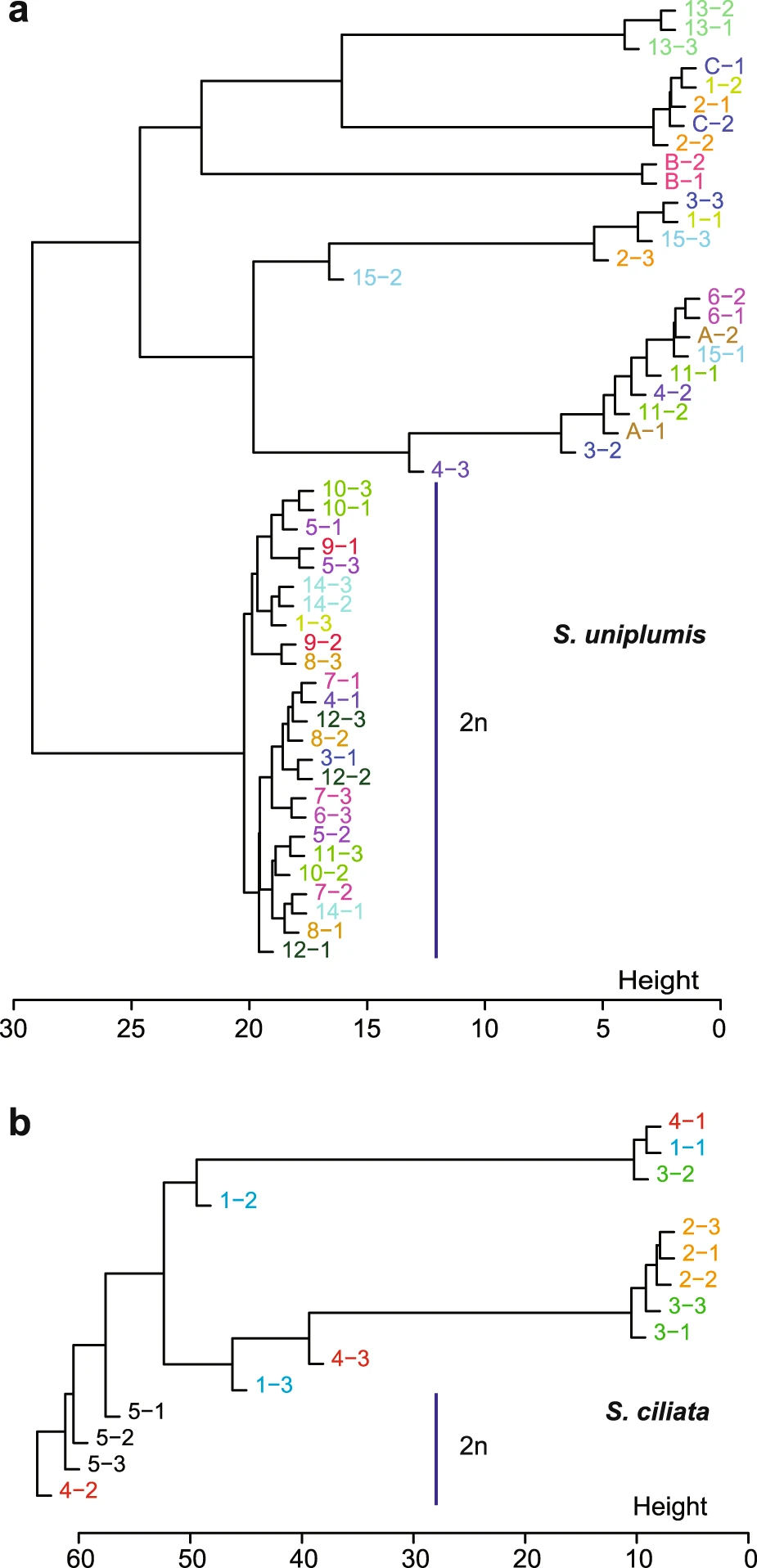
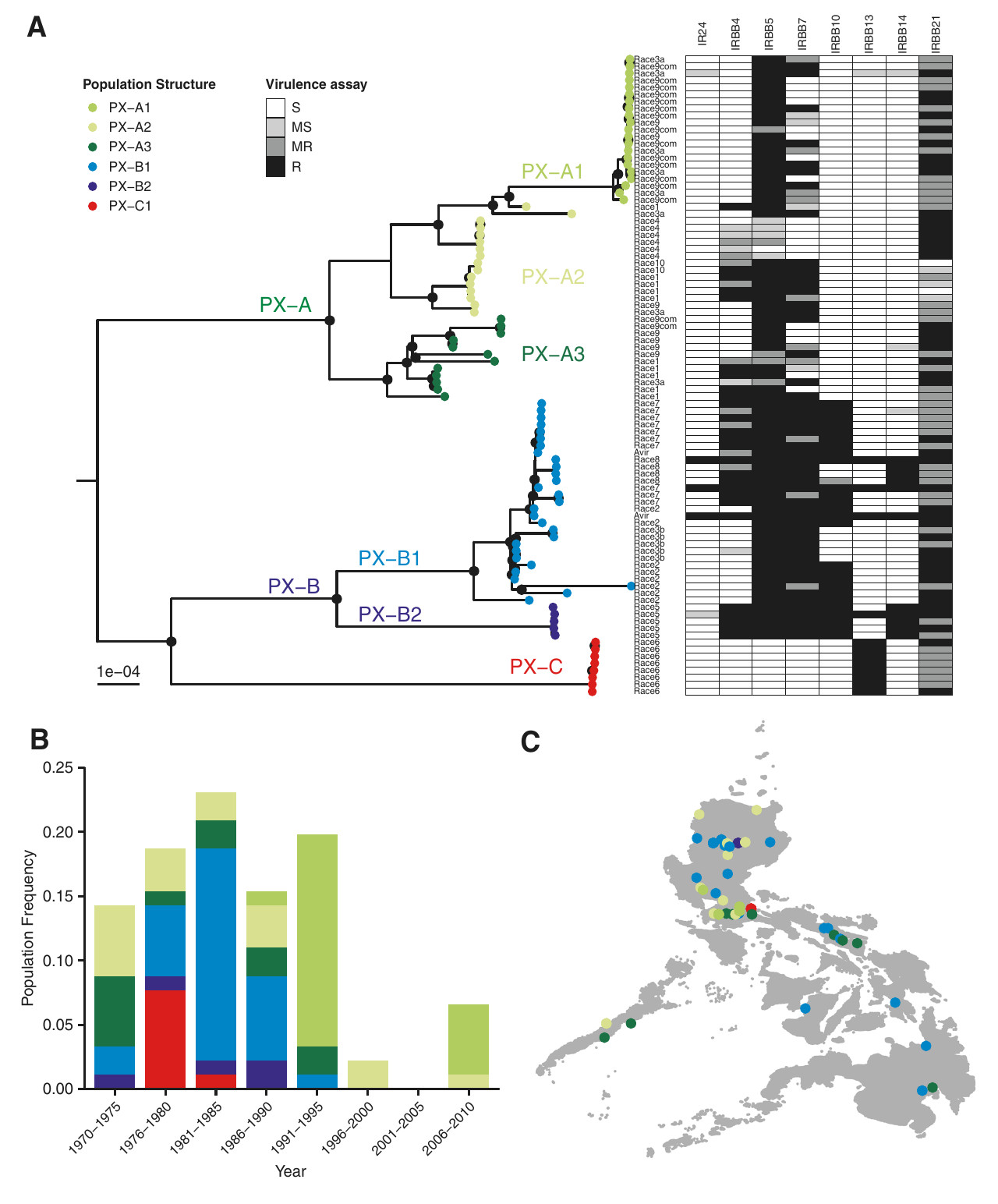
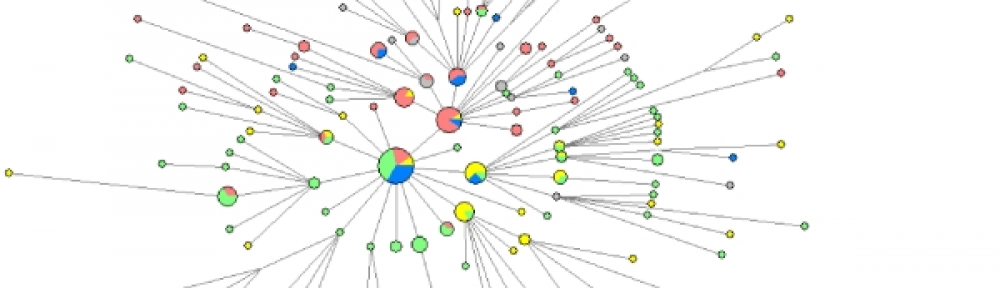
Relations d’apparentement entre les individus échantillonnés à la périphérie des cercles de fées namibiens des espèces Stipagrostis uniplumis (a) et S. ciliata (b). Il s’agit d’une étude de regroupement hiérarchique sur la base de la caractérisation moléculaire des individus. Les individus provenant du même cercle de fées sont indiqués par le même préfixe (chiffre avant le tiret) et la même couleur. Les individus avec un suffixe 1 et 2 sont échantillonnés à proximité dans chaque cercle de fées tandis que l’individu avec un suffixe 3 est échantillonné à l’opposé des deux précédents, par rapport au cercle de fées. Si certains individus d’un même cercle de fées présentent peu de variation génétique (ex : 2-1, 2-2 et 2-3 pour S. ciliata), d’autres sont très distincts génétiquement (3-1, 3-2 et 3-3 pour S. uniplumis). On observe aussi une nette distinction entre les individus diploïdes (notés 2n) et les autres individus, tétraploïdes.
L’étude a porté sur deux régions désertiques de Namibie, dans lesquelles les cercles de fées sont formés par Stipagrostis ciliata (5 cercles étudiés avec 3 plantes échantillonnées dans chaque cercle) et S. uniplumis (idem mais pour 15 cercles) respectivement. Le génotypage à plus de 60 000 portions du génome des plantes montre que les plantes constituant un cercle de fées ont des génomes différents. Mieux, elles sont parfois de niveaux de ploïdie différents : des plantes diploïdes et des plantes tétraploïdes peuvent pousser dans le même cercle. Le coefficient de consanguinité F calculé pour les échantillons diploïdes de S. uniplumis est de -0.025, c’est-à-dire très proche de 0, indiquant des plantes parfaitement allogames.
On peut donc exclure que les cercles de fées de Namibie résultent du développement végétatif d’un seul clone. Au contraire, les cercles de fées sont constitués de plusieurs génotypes distincts, probablement apparus par semis de graines issues d’allo-fécondations. Si on remet cela dans le contexte du suivi des plantes et globalement des cercles de fées au cours du temps, cela signifie un très grand décalage entre des cercles de fées qui peuvent perdurer des centaines d’années et des plantes individuelles, non clonales, qui ont une durée de vie courte, jusqu’à une dizaine d’année, généralement moins du fait d’événements de sécheresse.
Pourquoi des individus, en compétition pour les ressources hydriques, s’organiseraient en cercle ? Pourquoi ces cercles seraient aussi réguliers en diamètre et aussi stables dans le temps, sur des centaines d’années d’alternance de périodes sèches et de périodes humides, chez des individus avec des traits d’histoire de vie aussi peu stables qu’une reproduction sexuée et un temps intergénérationnel court ? L’absence de clonalité chez les plantes constituant les cercles de fées namibiens déconstruit les principales hypothèses proposées, donne matière à penser aux écologues comme aux mathématiciens et remystifient les cercles de fées aux yeux des naturalistes. Quadrature du cercle pour les uns et féérie pour les autres.
Références de l’article