Le sexe, c’est important, mais est-ce vital ? On a généralement pensé que oui, du fait que l’extrême majorité des Eucaryotes se reproduisent par voie sexuée. La reproduction sexuée apporterait en effet un avantage pour échapper aux parasites, en augmentant la diversité génétique dans les populations, et rendrait plus improbable le fait que le parasite puisse infiltrer les défenses de l’hôte. La reproduction sexuée permettrait aussi de se débarrasser des mutations faiblement délétères (le fardeau génétique) en générant des recombinaisons entre les portions chromosomiques. Malgré tout, il existe des Eucaryotes à reproduction asexuée mais ces lignées sont généralement considérées comme des cul-de-sacs évolutifs. Toutes ? Non, car quelques groupes d’organismes résistent encore et toujours à ce paradoxe (un ‘scandale évolutif’, d’après John Maynard Smith) : les Crustacés ostracodes, les Acariens Oribatida, les insectes Phasmes du genre Timema et les Rotifères bdelloïdes. Toutes ces espèces sont supposées avoir évolué depuis une lignée évolutive qui refuse la sexualité depuis fort longtemps. Des dizaines de millions d’années pour les Rotifères bdelloïdes, pensait-on. Ainsi, sur les centaines de milliers d’individus observés chez l’espèce de bdelloïdes Adineta vaga, aucun mâle n’a jamais été observé. Mais les Rotifères bdelloïdes n’ont-elles pas quelque chose à cacher sur leurs aventures sexuelles passées et/ou actuelles ?
Rotifère bdelloïde de l’espèce Adineta vaga. La barre d’échelle représente 100 µm. (Source : Vakhrusheva et al. 2020)
Non, pas de sex tape en Super 8. Juste les travaux d’Olga A. Vakhrusheva et ses collaborateurs et collaboratrices russes et états-unien·ne·s, publiés dans le journal Nature Communications en décembre 2020. Cette équipe a comparé le génome de onze individus de l’espèce Adineta vaga, échantillonnés sur des mousses vivant sur des peupliers trembles (Populus tremula) en Russie. Mais les analyses de clonalité ont été réalisées sur huit génomes, sachant que les trois autres constituaient un groupe différencié qui pouvait biaiser l’analyse. Les résultats vont clairement montrer que l’hypothèse de clonalité stricte est à exclure.
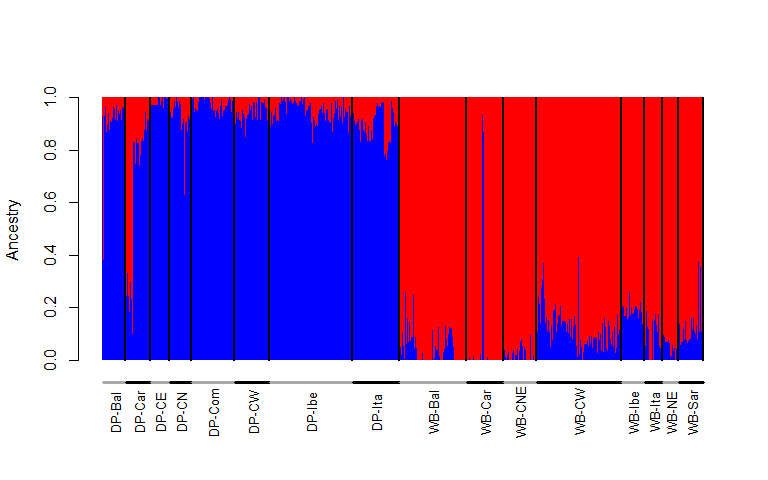
Un argument probant pour appuyer cette conclusion est l’observation d’une décroissance du déséquilibre de liaison (DL) avec la distance séparant les sites variables du génome. Le déséquilibre de liaison ou déséquilibre gamétique est la co-occurrence entre les allèles de deux loci (positions dans le génome) présents sur un même chromosome. En cas de clonalité, les mutations se sont accumulées au fur et à mesure dans les génomes des ancêtres d’un individu, entraînant ces co-occurrences. En cas de reproduction sexuée, les méioses (divisions cellulaires à l’origine des gamètes) font l’objet de recombinaisons entre chromosomes homologues. Ainsi, les co-occurrences qui ont pu se former entre des allèles à différents loci sont atténuées par des échanges de segments chromosomiques et ce phénomène est d’autant plus marqué que la distance séparant les loci est grande. Les données observées chez A. vaga ont été comparées à des données simulées en cas de stricte clonalité (stabilité du DL) ou en cas de reproduction sexuée (décroissance du DL avec la distance). Du fait de la décroissance du DL observée sur son génome, Adineta vaga montre clairement un profil incompatible avec une stricte clonalité.
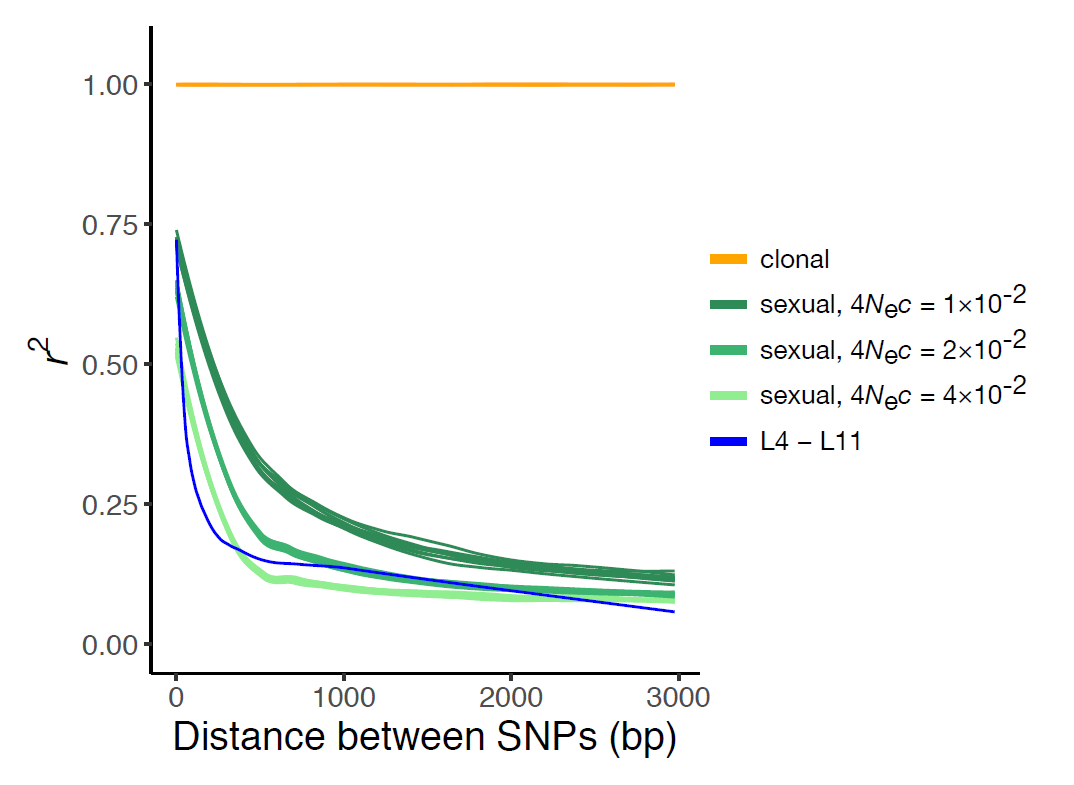
Décroissance du déséquilibre de liaison (DL), estimé par l’indice r², en fonction de la distance entre les variants nucléotidiques (Single Nucleotide Polymorphisms ou SNPs). En cas de stricte reproduction clonale, des simulations prédisent que le DL est maximal et stable (courbe orange). En cas de reproduction sexuée, des simulations prédisent que le DL décroît rapidement avec la distance (courbes vertes). Les nuances de vert représentent différentes valeurs du taux de recombinaison populationnel 4Nec, avec Ne l’effectif efficace de la population et c le taux de recombinaisons par nucléotide et par génération. La courbe bleue représente la décroissance réellement observée sur les données des 8 génomes de Adineta vaga analysés.
Cependant, avant de conclure définitivement à une reproduction sexuée chez ces Rotifères bdelloïdes, l’équipe de recherche a souhaité vérifier que les résultats ne pouvaient pas être expliqués par d’autres mécanismes. En premier lieu, l’action concertée des mutations et des conversions géniques. La conversion génique est le transfert d’ADN intra-individuel entre deux copies homologues du génome et peut se faire avec ou sans recombinaison. Pour distinguer d’une part les haplotypes (combinaisons d’allèles sur un même chromosome) générés par mutation puis conversion génique sans recombinaison et d’autre part les haplotypes générés par recombinaison durant les phases de reproduction sexuée, on a recherché spécifiquement les cas où les quatre haplotypes possibles AB, Ab, aB et ab étaient présents chez deux individus différents. La présence de ces quatre haplotypes chez seulement deux individus est en effet une bonne signature de l’action de la recombinaison homologue. Ces haplotypes recombinants existent bel et bien dans le génome de Adineta vaga et sont d’autant plus fréquents que la distance entre les sites variables comparés augmente, en cohérence avec l’augmentation de la fréquence des recombinaisons avec la distance.
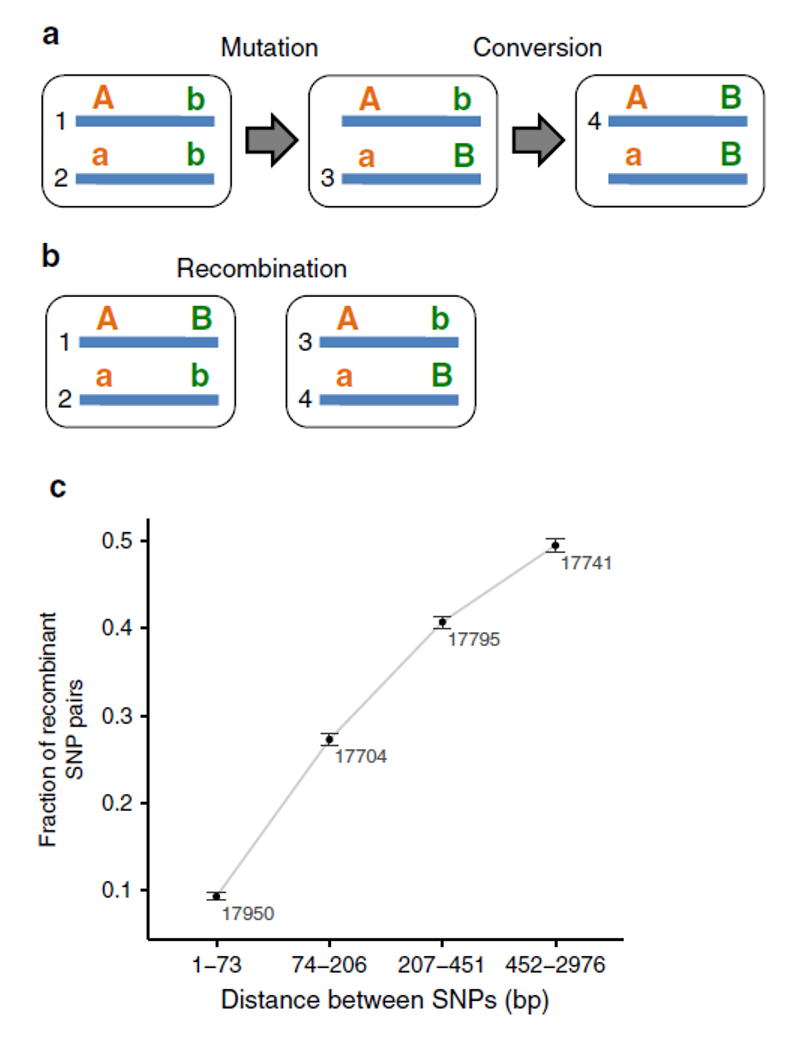
Confirmation de l’implication de recombinaisons homologues plutôt que de la conversion génique. (a) Mécanisme expliquant l’apparition de quatre haplotypes (notés de 1 à 4) à partir d’une mutation (b muté vers B) puis d’un événement de conversion génique (B copié sur b). Les quatre haplotypes se retrouvent dans trois organismes différents et le resteront si les organismes se reproduisent de manière clonale (sauf nouvelle mutation). (b) A partir de deux haplotypes AB/ab, un événement de recombinaison homologue peut produire deux gamètes recombinés Ab et aB. Ceux-ci peuvent finir par se retrouver dans un même individu, par fécondation. (c) Pourcentage d’haplotypes recombinants en fonction de la distance entre les SNPs constituant l’haplotype. On observe que la proportion d’haplotypes recombinants augmente avec la distance : elle passe de 10% pour des SNPs distants de moins de 74 paires de bases à 50% pour des SNPs distants de plus de 451 paires de bases.
Toutefois, il reste la possibilité que ces recombinaisons homologues se soient passées, non pas suite à des échanges inter-individuels (fécondation à partir de gamètes issus de la méiose), mais suite à des recombinaisons mitotiques à l’intérieur d’un même organisme en absence d’échanges inter-individuels. Afin d’exclure cette dernière hypothèse, l’équipe a caractérisé le coefficient de consanguinité FIS = 1 – Ho/He, avec Ho comme le taux d’hétérozygotes observé et He le taux d’hétérozygotes attendu sous l’hypothèse d’équilibre de Hardy-Weinberg. L’équilibre de Hardy-Weinberg est un modèle théorique fondateur de la génétique des populations et repose sur huit hypothèses dont celle d’une reproduction sexuée. Ainsi, si la population biologique étudiée est à l’équilibre de Hardy-Weinberg, le taux d’hétérozygotes observé Ho est identique au taux d’hétérozygotes attendu He, ce qui entraîne un FIS nul. En cas de reproduction clonale, les mutations sont accumulées à chacun des deux allèles indépendamment, ce qui entraine l’observation d’un excès d’hétérozygotes, et par conséquent un FIS négatif. Des simulations montrent que jusqu’à 90% de taux de clonalité (1 reproduction sexuée tous les 10 cycles de reproduction), les valeurs de FIS restent proches de zéro. Toutefois, quand les reproductions sexuées viennent à se raréfier davantage, le FIS chute vers des valeurs négatives, comme le prévoit la théorie. Les sites variables détectés chez Adineta vaga montrent des valeurs de FIS proches de zéro et sont donc beaucoup plus en accord avec l’hypothèse de reproductions sexuées régulières plutôt qu’avec une reproduction clonale.
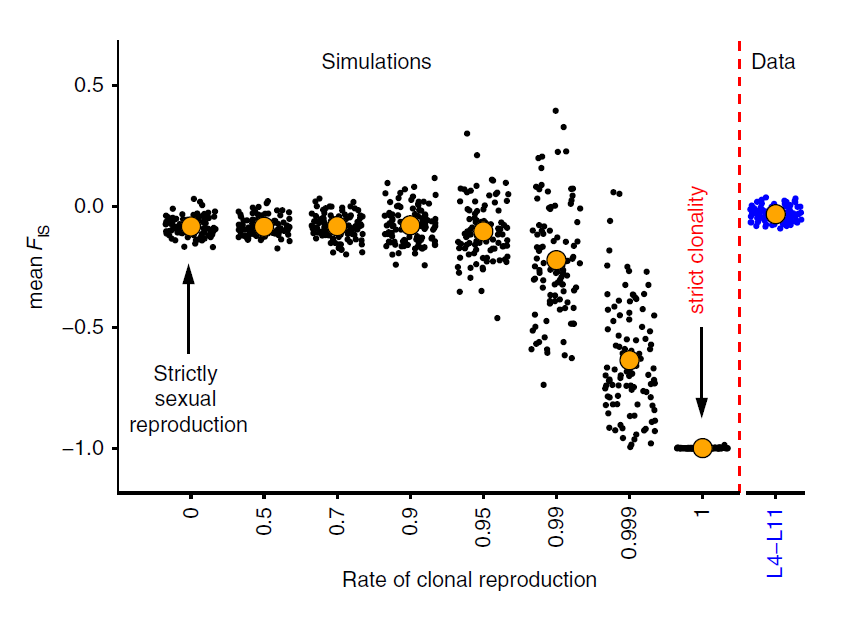
Coefficient de consanguinité FIS en fonction du taux de reproduction clonale. Les points noirs représentent des FIS calculés sur des données simulées selon le taux de clonalité, tandis que les points bleus représentent des FIS calculés sur des données observées chez les 8 génomes de Adineta vaga. Les gros points orange sont les moyennes pour chaque groupe de points.
On a donc vérifié que le génome de ces Rotifères bdelloïdes avait été fortement influencé par des événements de recombinaison et par des échanges inter-individuels. Reste à déterminer quel type d’échanges inter-individuels parmi les deux mécanismes suivants : la reproduction sexuée méiotique (un classique !) et le transfert de gènes horizontal (HGT). Il a en effet été montré dans de précédents articles que les Rotifères bdelloïdes ont aussi largement pratiqué les transferts horizontaux (incorporation dans son propre génome d’ADN exogène) à partir de différentes espèces. Cependant, la recombinaison méiotique conventionnelle implique l’appariement de chromosomes homologues. Or, chez les Rotifères bdelloïdes, il existe bien des régions homologues entre chromosomes mais celles-ci sont assez discontinues, ce qui laisse penser que la méiose conventionnelle est peut-être compromise, même s’il existe un vif débat sur le sujet. Chez certaines plantes, dont les plantes du genre Oenothera (appelées aussi herbes aux ânes ou onagres), et probablement certains animaux, il existe un type de méiose non conventionnelle, dit ‘de type Oenothera‘, qui fonctionne sans nécessité de paires de chromosomes homologues : pendant la méiose, les chromosomes paternels et maternels sont alternés à la queue leu-leu, formant un cercle de chromosomes. Chaque lot de chromosomes paternel ou maternel est ensuite transmis à des gamètes différents. La phylogénie de différentes régions du génome de Adineta vaga a été réalisée pour clarifier le mode de transmission d’information génétique entre individus. De nombreuses incongruences ont été identifiées : pour un même individu, chaque membre d’une paire d’haplotypes avait souvent comme plus proche voisin dans la phylogénie des haplotypes provenant de différents individus. Par exemple, l’haplotype L6.1 (1er haplotype de l’individu L6) peut se retrouver proche dans la phylogénie de l’haplotype L7.1 alors que l’haplotype L6.2 (2e haplotype de l’individu L6) se retrouve proche de L9.2 au lieu de L7.2. Ces incongruences témoignent d’échanges génétiques entre individus. Une analyse plus fine sur l’ensemble du génome montre que les patrons d’incongruence sont très diversifiés et sont plus en faveur d’échanges génétiques inter-individuels par méiose conventionnelle ou par transfert de gènes horizontal plutôt que par méiose de type Oenothera.
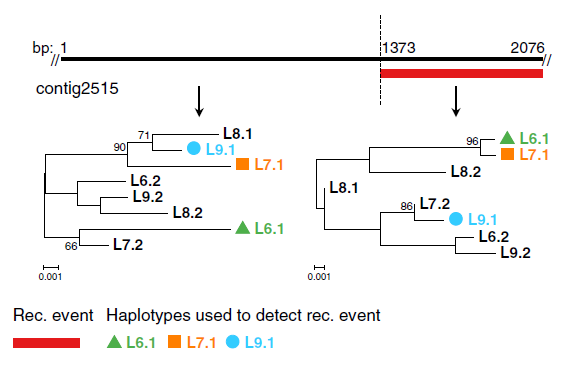
Exemple d’incongruences sur la phylogénie de chacun des deux haplotypes des quatre individus L6 à L9. Ici, deux régions nucléotidiques délimitées par la ligne pointillée ont été utilisées pour construire chacun des arbres phylogénétiques. Les indices 1 et 2 après le nom de l’individu désigne le numéro de l’haplotype. Seuls les haplotypes dont la reconstruction est la plus fiable (écrits en couleur) ont été utilisés pour tester l’existence d’une recombinaison entre ces deux régions. L’existence de deux types d’incongruence à deux régions adjacentes suggère un événement de recombinaison entre les deux régions analysées.
Il est difficile de trancher entre les deux hypothèses finales de méiose conventionnelle et de transferts de gènes horizontaux. Toutefois, les auteurs suggèrent que les trois individus laissés de côté au début de l’étude seraient des hybrides entre le groupe d’individus étudiés dans l’article et une autre population non échantillonnée ici. Ainsi, cela accréditerait plutôt l’hypothèse de reproduction sexuée par méiose conventionnelle.
Si les résultats vont dans le sens de fréquents événements de reproduction sexuée chez cette espèce qu’on considérait dériver d’une lignée ayant délaissé les reproductions sexuées depuis plusieurs dizaines de millions d’années, il n’en demeure pas moins que ces événements de reproduction sexuée ne seraient pas aussi fréquents que les événements de reproduction asexuée. Les auteurs estiment que les patrons de recombinaison observés seraient compatibles avec une fréquence de 1 méiose toutes les 10 à 100 générations, ce qui est encore beaucoup chez une espèce où aucun mâle n’a jamais été observé ! L’hypothèse de transfert de gènes horizontal (HGT) est davantage compatible avec l’absence d’observation de mâle, mais pas avec l’observation d’individus hybrides et elle requerrait une fréquence élevée de 1 HGT toutes les 1 à 10 générations.
Au final, on peut conclure que cette espèce de Rotifère bdelloïde présente de nombreuses recombinaisons dans son génome qui montrent clairement qu’elle use de mécanismes qui permettent un brassage de la diversité génétique. Les mécanismes utilisés ne sont pas encore clairement tranchés entre reproduction sexuée et transferts de gènes horizontaux. Mais la démonstration de brassage génétique fréquent chez cette espèce à reproduction (au moins majoritairement) clonale permet d’étouffer ce “scandale évolutif” d’une lignée évolutive sans avenir (cul-de-sac évolutif) mais qui perdure depuis des dizaines de millions d’années. Reste à trouver la pièce à conviction démontrant le mécanisme : sex tape ou autre…
Références de l’article