La limitation de la reproduction entre les individus jugés inaptes est l’un des fondements de l’eugénisme. Ainsi, l’inventeur du téléphone Alexander Graham Bell, qui fut aussi éducateur dans des écoles pour personnes sourdes et marié à une femme sourde, expose ses craintes en 1883 dans une publication intitulée ‘Memoir Upon the Formation of a Deaf Variety of the Human Race’. Il y explique que les mariages très fréquents entre personnes sourdes, encouragés à la fois par le déficit de communication entre personnes entendantes et personnes sourdes et par le développement de structures éducatives dédiées aux sourds, risquait à terme de créer une sous-population humaine sourde. A la demande de Bell, une étude est menée dans les années 1880 par Edward Allen Fay sur 4 471 pedigrees de sourds étudiant au Gallauget College (une Université dédiée aux sourds et malentendants, à Washington DC) ou inscrits dans des écoles de sourds des Etats-Unis. Les conclusions de cette étude démentent les craintes de Bell : les mariages entre sourds n’augmentent presque pas la probabilité de naissance d’un enfant sourd.

Le comédien et metteur en scène Levent Beskardes signant le mot “poème” pour la série Les Mots du silence, de la photographe Jennifer Lescouët (Jennifer Lescouët, CC-BY-SA 4.0).
Le déterminisme génétique de la surdité, inconnu au XIXe siècle, est aujourd’hui mieux documenté. La surdité congénitale est causée dans un quart des cas par des allèles déficients récessifs du gène de la connexine 26 (GJB2), mais ce sont des mutations sur plus de 140 gènes qui ont été rapportées. Ainsi, des parents sourds peuvent être homozygotes pour des allèles déficients mais à des gènes différents ; leurs enfants, parfaitement entendants, bénéficieront d’un effet de complémentation en récupérant un allèle fonctionnel pour chacun des gènes défectueux chez leurs parents. Une autre cause de naissance d’enfants entendants issus d’un couple de sourds est que la surdité de certains parents n’a pas toujours une origine génétique, mais parfois une cause environnementale, comme par exemple des infections.
Le fait qu’une personne sourde soit plus susceptible de fonder une famille avec une autre personne sourde est considéré comme une forme d’homogamie linguistique. En biologie, l’homogamie est définie comme un choix de partenaire de reproduction en direction des individus présentant des similitudes phénotypiques avec soi. Au début du XXe siècle, les travaux des biostatisticiens Ronald Fisher et Sewall Wright sur les attendus théoriques de l’homogamie en génétique des populations sont éclairants sur ce sujet. Dans le cas théorique d’un gène unique à transmission récessive contrôlant la maladie, des parents atteints donneront systématiquement des enfants atteints. Ce modèle théorique prédit que la proportion d’homozygotes augmenterait dans la population et par conséquent le taux de personnes sourdes. Toutefois, la fréquence de l’allèle récessif déficient resterait constante.
Néanmoins des études récentes, réalisées sur la population américaine contemporaine, suggèrent que les unions entre personnes sourdes ont entrainé l’augmentation de la prévalence de la surdité dans la population, mais aussi de la fréquence des allèles responsables. De tels résultats peuvent avoir des impacts négatifs sur le financement de structures éducatives accusées de renforcer l’endogamie dans la communauté des sourds. Derek C. Braun et ses collaborateurs américains de la Gallaudet University ont cherché à tester ces conclusions par des simulations bioinformatiques incluant des paramètres choisis au regard de la littérature disponible sur ce handicap. Leur démarche et leurs résultats ont été publiés dans la revue PLoS ONE en novembre 2020.
C’est ainsi l’évolution d’une population de 200 000 individus qui a été simulée sur 20 générations, soit environ 400 ans, c’est-à-dire l’âge approximatif de la langue des signes d’après l’article. La fréquence initiale de l’allèle récessif de la surdité a été fixée à 1,304%, c’est-à-dire la fréquence de l’allèle c.35delG du gène GJB2, majoritaire chez les Américains blancs et les Européens. Une proportion de 0,8‰ des individus sains ont été rendus sourds, ce qui correspond à la proportion des cas de surdité causés par d’autres gènes, par des causes épigénétiques ou par des troubles d’origine périnatale. Quelle que soit la cause de leur surdité, les personnes sourdes ont été accouplées, avec un taux d’homogamie linguistique variable, sachant que dans la population contemporaine réelle, ce taux d’homogamie est estimé à 90%.
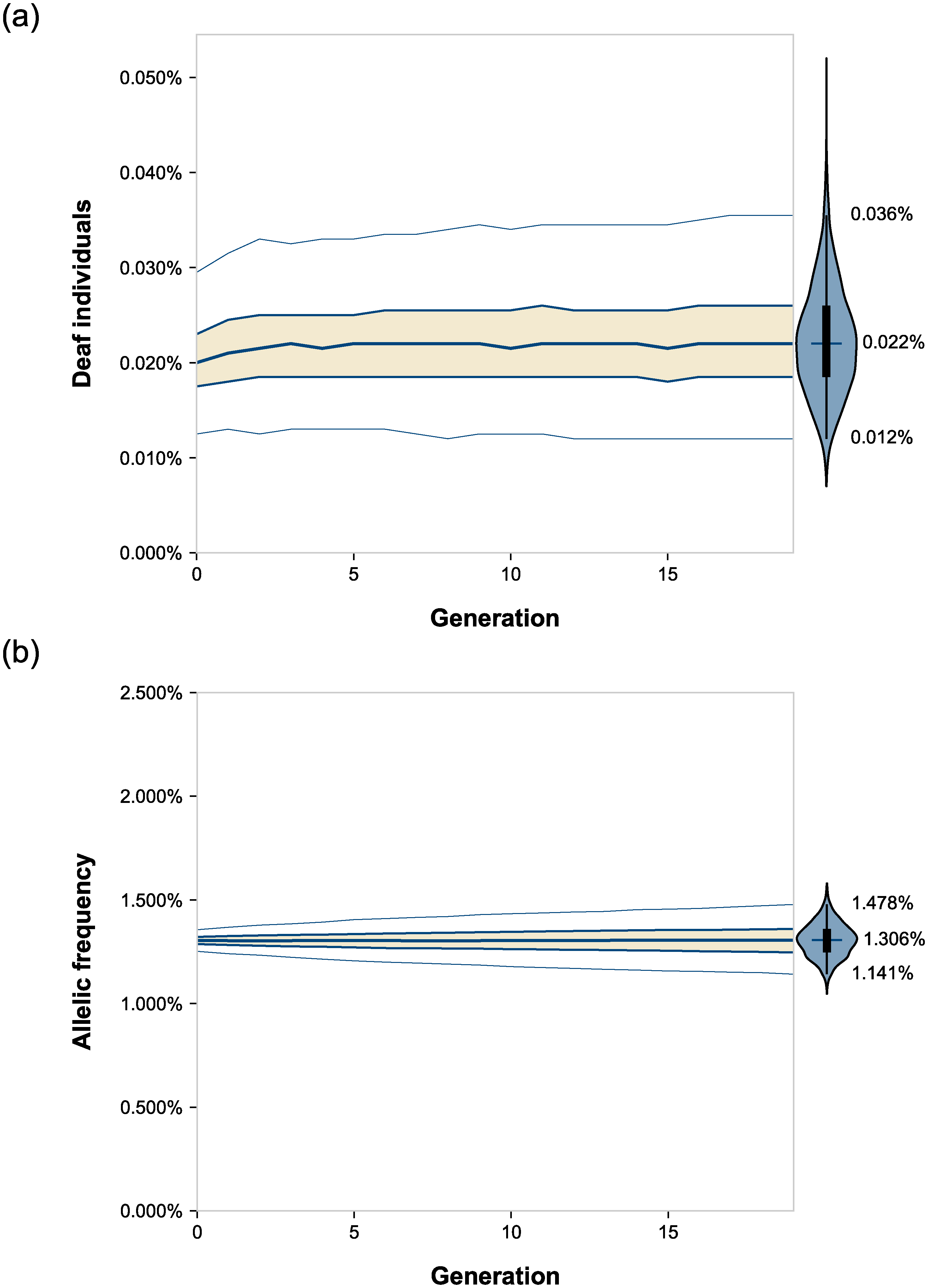
Effet de l’homogamie sur (a) la prévalence de la surdité et (b) la fréquence de l’allèle récessif à l’origine de la surdité, au cours de 20 générations. Les courbes représentent des statistiques descriptives de la dispersion des valeurs selon les simulations avec de haut en bas : le 98e percentile, le 3e quartile, la médiane, le 1er quartile et le 2e percentile. Cette dispersion est aussi visible sous forme d’un diagramme en violon, sur la droite. Ces simulations ont été réalisées avec une valeur sélective relative pour les sourds de 1.
Après simulation des 20 générations, la fréquence des sourds est passée à 0,022% avec un taux d’homogamie de 90% contre 0,017% sans homogamie, soit une augmentation significative de 23% imputable à l’homogamie. Les simulations montrent que c’est au cours des trois premières générations d’homogamie que cette variation s’opère puis les variations sont marginales sur les générations suivantes. Au contraire, la fréquence de l’allèle récessif n’a pas varié significativement : 1,306% avec homogamie contre 1 ,304% sans homogamie. Ces simulations informatiques sont donc en accord avec les prédictions théoriques basées sur des équations mathématiques.
L’étude va plus loin en simulant l’impact synergique de l’homogamie et d’une hausse de la valeur sélective relative des individus sourds, c’est-à-dire de leur capacité à survivre et à se reproduire comparativement aux individus normaux. En supposant notamment une valeur sélective relative des sourds de 1,5, c’est-à-dire une augmentation de 50% par rapport à celle des personnes entendantes, voire plus, on observerait une augmentation significative de la fréquence de l’allèle récessif déficient dans la population. Mais cette valeur relative est loin d’être en phase avec la réalité puisque la fertilité relative des personnes sourdes est généralement faible, comprise entre 0,31 et 0,91 selon les études, c’est-à-dire toujours une fertilité plus faible que celle des personnes entendantes.
Avec cette fertilité réduite, comment expliquer que certains allèles récessifs déficients aient quand même été mesurés entre 1% et 4,4% dans les populations humaines ? Une des hypothèses avancées est celle d’une sélection équilibrante pour ces allèles. La fréquence allélique actuelle serait le résultat d’un équilibre entre l’effet négatif lié à la fécondité réduite observée chez les personnes sourdes et un effet positif que pourraient apporter ces allèles. Il a notamment été montré que le gène GJB2 est requis pour l’infection de Shigella flexneri, agent bactérien de la shigellose, une forme de dysenterie. Pour vérifier si les allèles déficients des sourds entraînent réellement une résistance à la dysenterie et si cette résistance s’exprime à l’état hétérozygote autant qu’à l’état homozygote, il faudra prêter l’oreille aux futurs travaux scientifiques.
Références de l’article