* A l’attention de l’historien du 4e millénaire qui aura eu la folie de choisir comme sujet de thèse “L’humour raté dans les blogues de génétique des populations au 3e millénaire”, l’auteur de ce billet précise qu’il a été rédigé en pleine période d’épidémie de Covid-19 de 2020 (la première année de l’épidémie) où la Solution Hydro-Alcoolique (SHA) était particulièrement recherchée pour ne pas tomber malade comme un chien.
Si la domestication du chien a commencé il y a 15 000 ans, le concept de races homogènes apparaît au 19e siècle et on distingue aujourd’hui 400 races de chiens. On a déjà évoqué sur ce blogue les spécificités génétiques des races modernes de chiens qui ont subi une sélection drastique des individus reproducteurs répondant aux standards de la race. La réduction de l’effectif efficace et l’augmentation de la consanguinité à l’intérieur de la race ont abouti à des races bien différenciées génétiquement, avec une perte de diversité génétique et l’accumulation d’allèles délétères chez de nombreuses races (voir aussi la sélection moderne des races équines qui a eu des effets similaires), causant des maladies génétiques fréquentes chez les races modernes. Toutefois, les races canines sont-elles réellement si homogènes génétiquement ? Ou est-on capable de distinguer plusieurs sous-populations distinctes chez des races qui, malgré une standardisation, se retrouvent mondialisées (sélectionnées sur plusieurs continents) et peuvent avoir plusieurs usages avec des schémas de sélection parfois indépendants ?

Six races de chiens étudiées dans l’article : la Levrette d’Italie (Chrsitina, CC-BY 2.0), le Shetland (Sannse, CC-BY-SA 3.0), le Lévrier anglais en pleine course (Matt Schumitz, CC-BY-SA 4.0), le Labrador Retriever guide d’aveugle (Honza Groh, CC-BY-SA 3.0), le chien finnois de Laponie (Svenska Mässan, CC-BY 2.0) et les types de Bergers belges : le Groenendael (Tsaag Valren, CC-BY-SA 4.0), le Malinois (Caronna, CC-BY-SA 3.0), le Tervueren (Ulrik Fällstrom, CC-BY-SA 2.5) et le Laekenois (Sannse, CC-BY-SA 3.0). Toutes les photos sont issues de Wikimedia Commons.
L’article de Sara Lampi et de ses collaborateurs finlandais, paru le 9 juin 2020 dans la revue Canine Medicine and Genetics, caractérise la structuration génétique intra-race de six races de chiens. Ces six races n’ont pas été choisies au hasard, mais en fonction de différentes caractéristiques :
- Variation d’usage : la Levrette d’Italie (ou petit lévrier italien) et le Lévrier anglais (ou lévrier greyhound) sont des chiens utilisés pour les compétitions de courses de vitesse, tandis que le Labrador (ou Labrador Retriever) est utilisé comme chien de service, notamment guides d’aveugle, sachant que des individus de toutes ces races sont aussi élevés comme des chiens de compagnie ou pour les concours canins.
- Variation de popularité : si le Shetland (ou berger des Shetland) présente une popularité internationale, le Chien finnois de Laponie est peu connu en dehors de la Finlande.
- Variation de types : si elles sont toutes regroupées sous la race Berger Belge, on en distingue quatre variétés : le Malinois, le Groenendael, le Tervueren et le Laekenois.
Entre 90 et 608 individus de chaque race ont été comparés pour 1 319 régions ponctuelles du génome (Single Nucleotide Polymorphisms) et la structuration intra-race a été étudiée par des approches de génétique des populations.
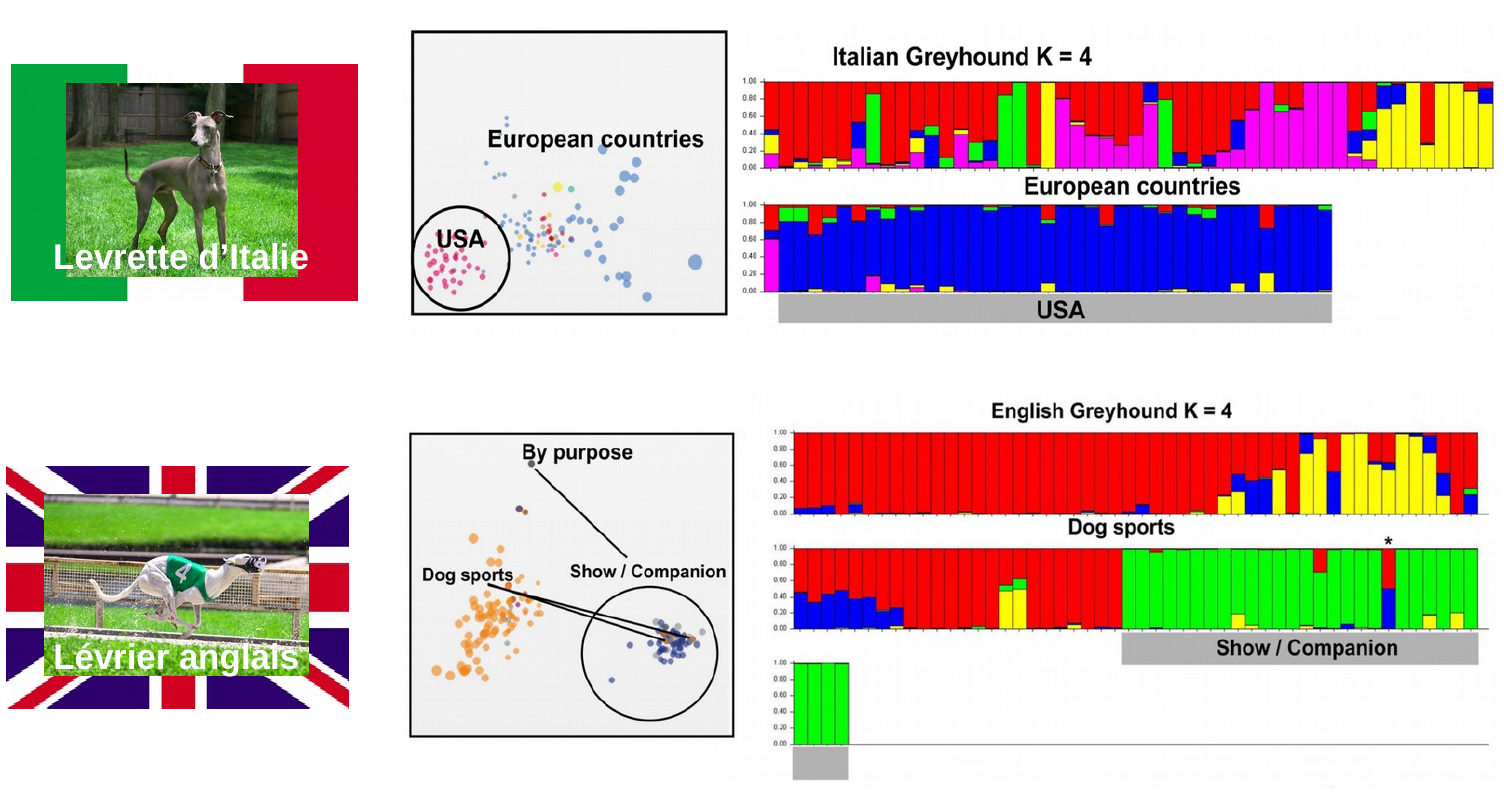
Structuration génétique selon l’origine géographique pour la levrette d’Italie (en haut) et selon l’usage pour le lévrier anglais (en bas). La structuration de la diversité génétique intra-race est étudiée par deux méthodes : le positionnement multidimensionnel (ou multimensional scaling, MDS), au centre, et la méthode d’inférence bayésienne STRUCTURE, à droite. Pour la méthode MDS, les points représentent des individus de la race, colorés en fonction de l’origine géographique (levrette d’Italie) ou en fonction de l’usage (lévrier anglais). La position des points symbolise la proximité génétique des individus. On distingue un nuage de points à part pour les levrettes d’Italie américaines (rose) : cela signifie qu’elles sont génétiquement distinctes des individus européens (autres couleurs). De même, les lévriers anglais utilisés pour les courses de vitesse (orange) sont génétiquement distincts des individus utilisés comme chiens de compagnie ou pour les concours de beauté (bleu). Des traits indiquent de rares individus dont le fond génétique est en désaccord avec leur usage. Sur les analyses de STRUCTURE (à droite), les groupes génétiques inférés par l’analyse des marqueurs SNPs sont représentés par des couleurs différentes. Pour chaque race, c’est la solution à K=4 groupes qui semblait la plus vraisemblable. Les individus sont représentés par les bâtons de l’axe des abscisses tandis que les ordonnées représentent la probabilité d’assignation d’un individu à l’un des quatre groupes génétiques. Ces résultats de STRUCTURE viennent compléter les analyses de MDS. Pour la levrette d’Italie, les chiens des USA forment un ensemble homogène, assigné majoritairement au groupe génétique bleu, ce qui traduit leur diversité génétique réduite. Aucun individu européen n’est assigné majoritairement à la population bleue, ce qui traduit l’isolement des Américains vis-à-vis des Européens. Les grandes différences d’assignation des individus européens montre la diversité plus importante des levrettes d’Italie européennes, en cohérence avec l’origine européenne de la race. L’analyse STRUCTURE pour les lévriers anglais confirme aussi la distinction génétique selon les usages : les chiens de concours de beauté ou de compagnie sont majoritairement assignés au groupe génétique vert, tandis que les chiens de course sont majoritairement assignés aux groupes rouge, jaune ou bleu.
L’origine géographique est le facteur explicatif principal de la structuration génétique pour la Levrette d’Italie et le Shetland. Pour chaque race, les chiens américains semblent représenter un sous-échantillon des chiens européens. Par exemple, pour la levrette d’Italie, l’indice de fixation FST est plus grand chez l’échantillon des USA (FST=0,15) par comparaison de celui de l’échantillon européen (FST=0,08). Les individus utilisés pour la reproduction sont probablement volontiers échangés entre pays européens, dont ces races sont originaires et pour lesquels la diversité est la plus forte. Au contraire, les échanges transatlantiques sont probablement plus rares. Enfin, les individus américains seraient le résultat d’un effet de fondation, c’est-à-dire d’une réduction de la diversité consécutivement à la “colonisation” d’un nouvel espace par un sous-échantillon de la population originelle, européenne.
L’usage est le facteur explicatif majeur de la structuration génétique pour le Lévrier anglais et le Labrador. Les Lévriers anglais utilisés pour les courses se distinguent génétiquement de ceux utilisés pour les concours de beauté ou comme chiens de compagnie. Un résultat similaire est trouvé entre les Labradors utilisés comme chiens de services et les Labradors de concours. Cela témoignerait de schémas de croisements relativement indépendants entre les usages, ce qui a mené à une dérive génétique au sein de chaque usage. Les chiens de concours (FST=0,34) sont largement plus différenciés que les chiens de course (FST=0,07), chez le Lévrier anglais (résultat similaire chez le Labrador), ce qui s’explique peut-être par la sur-représentation de quelques individus primés dans les pedigrees de ce type de chiens, entraînant une diminution de l’effectif efficace.
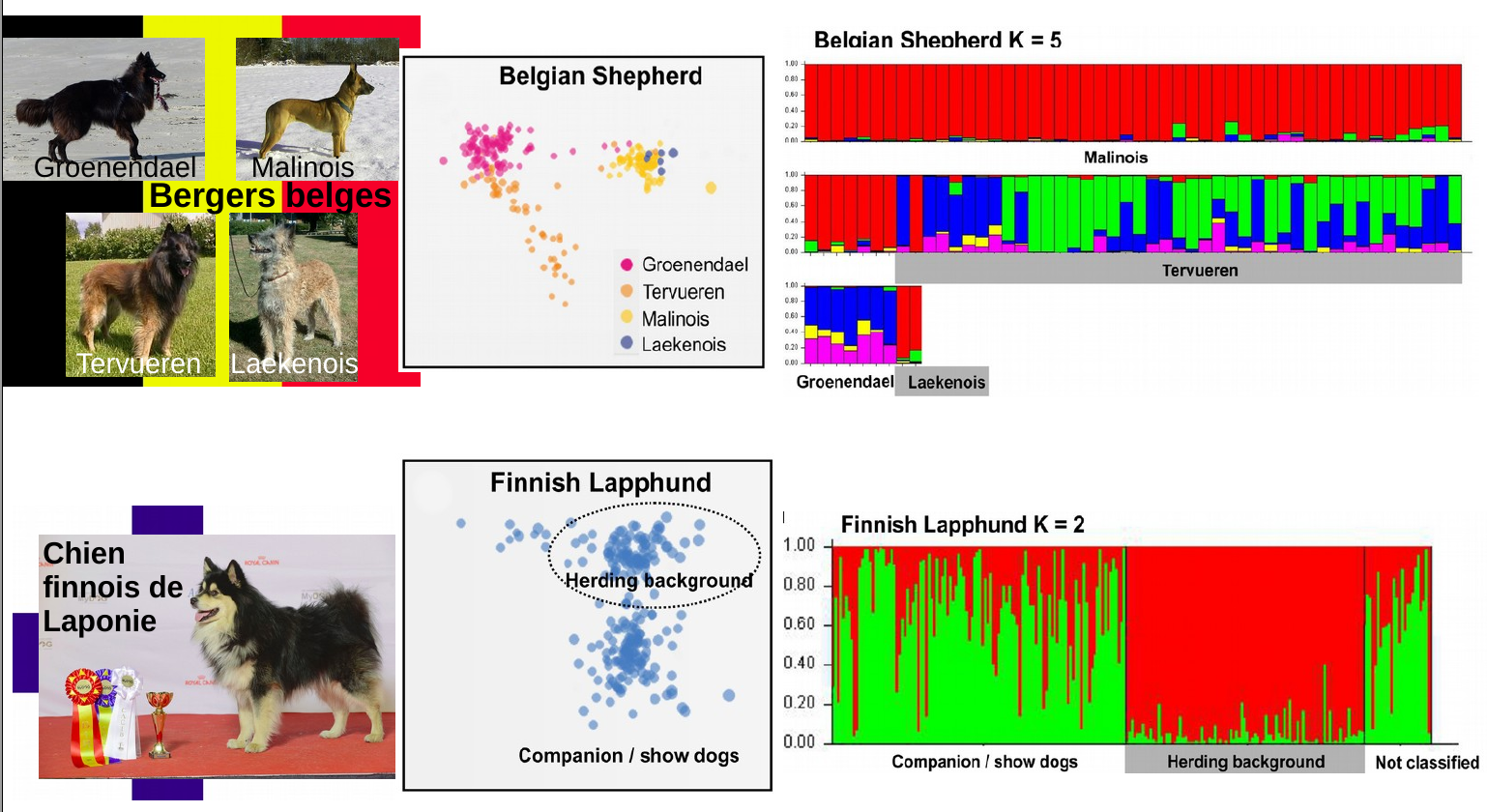
Structuration génétique pour les variétés morphologiques chez le Berger belge (en haut) et pour les stratégies de sélection chez le Chien finnois de Laponie (en bas). Pour les explications techniques sur les méthodes, voir la précédente figure. Les analyses montrent que les Malinois et les Laekenois ne sont pas génétiquement distincts, tout du moins pas sur les marqueurs SNPs répartis sur tout le génome : on pourrait probablement les distinguer en étudiant la variabilité génétique du gène expliquant l’aspect du poil, qui est le caractère phénotypique majeur distinguant ces deux variétés de Bergers belges. Les Groenendaels et les Tervuerens sont génétiquement distincts des deux premiers mais moins distincts l’un par rapport à l’autre, en cohérence avec leurs ressemblances phénotypiques (c’est principalement la couleur du pelage qui les distingue). Pour le Chien finnois de Laponie, les individus issus d’un schéma de sélection plus proche des chiens utilisés par les Samis, des éleveurs de rennes, (‘Herding background’) sont génétiquement distincts des autres individus de la race, comme le montrent la position des points sur la MDS et l’assignation majoritaire au groupe rouge par STRUCTURE, alors que les autres individus sont majoritairement assignés au groupe vert. L’article précise que des chiens issus de ce schéma de sélection proche des chiens de Samis sont régulièrement utilisés comme géniteurs dans les schémas de sélection classique (voir les individus majoritairement rouges, au milieu des individus verts). L’inverse est moins vrai (pas d’individu majoritairement vert au milieu des rouges).
L’analyse des variétés de Bergers belges montre que le Malinois et le Laekenois sont génétiquement différenciés du Groenendael et du Tervueren. Les deux premiers types partagent le point commun d’être particulièrement utilisés comme chiens de travail (bergers mais aussi comme chiens policiers) et se distinguent par le type de pelage (le Laekenois est à poils durs), qui est un caractère monogénique. Les croisements entre ces deux variétés sont autorisés, expliquant leur proximité génétique. Le Groenendael et le Tervueren se distinguent aussi pour un caractère monogénique : la couleur du pelage (noir chez le Groenendael et sable chez le Tervueren). Ainsi, dans une même portée où ce caractère ségrège, des chiots peuvent être inscrits dans l’une ou l’autre des variétés.
Deux groupes génétiques différenciés se distinguent pour les Chiens finnois de Laponie. L’analyse des pedigrees des individus a permis de comprendre les raisons de cette distinction. Même si les deux groupes génétiques sont phénotypiquement difficiles à différencier, le groupe désigné comme ‘Herding background’ correspond aux descendants de croisements menés par une association fondée en 1981 et souhaitant “revenir aux sources” de la race en repartant des chiens de berger utilisés par les Samis pour garder les rennes, l’utilisation originelle de la race.
Pour conclure, cette étude montre que malgré les standards définis pour chaque race, plusieurs sous-populations génétiquement différenciées peuvent être identifiées. La mondialisation des races de chiens les plus populaires a entraîné des effets de fondation en dehors de leurs zones d’origine et les migrations peu fréquentes entre les groupes géographiquement séparés contribuent à augmenter l’isolement génétique. L’élitisation de certains pedigrees qui donnent la part belle aux individus primés aux concours contribue aussi à entraîner une dérive génétique par rapport aux autres individus de la race utilisés comme chiens de travail. Enfin, des visions et des pratiques différentes de l’activité de sélection au sein d’une même race peut aussi entraîner une structuration de la diversité génétique. Dans un contexte où la baisse de diversité génétique chez des races modernes de plus en plus consanguines a multiplié les maladies génétiques, l’identification des populations structurées dans la race pourrait permettre d’atténuer cette consanguinité et de mieux raisonner la gestion de la diversité intra-race. Même si l’expression “malade comme un chien” trouve ses origines bien avant l’apparition des races modernes de chiens, ce progrès contribuerait peut-être à vider cette expression de son sens moderne.
Références de l’article