La domestication du cheval a révolutionné les civilisations humaines, aussi bien en terme de moyens de transport, d’échanges commerciaux ou de stratégies de guerre. Les premières traces de traite de jument, de harnachement ou de mise en captivité de cheval remontent à 5 500 ans dans les steppes d’Asie centrale. Cependant, les chevaux concernés ne seraient pas les ancêtres des chevaux modernes (Equus caballus) mais ceux des chevaux de Przewalski (Equus przewalskii). Une part de mystère subsiste quant au lieu de la domestication du cheval moderne : les steppes pontiques (au Sud-Est de l’Europe), l’Anatolie ou la péninsule ibérique ? Une précédente étude suggère aussi que suite à cette domestication, le génome du cheval aurait beaucoup changé au cours des 2 300 dernières années.

Reconstitution d’un cataphractaire sassanide. Les guerres entre les Sassanides (dynastie perse) et les Byzantins à partir du IVe siècle suivies des invasions arabes auraient contribué à introduire le cheval persan en Europe. Source : John Tremelling, GNU Free Documentation License, Wikimedia.
Antoine Fages, Kristian Hanghøj, Naveed Khan et leurs collaborateurs d’un large consortium international ont cherché à tester ces hypothèses dans un article publié dans le journal Cell, le 30 mai 2019. Ils se sont basés sur sur le génome de 30 chevaux modernes, les génomes anciens obtenus de 129 individus répartis sur les six derniers millénaires, plus des marqueurs génétiques à l’échelle du génome pour 149 autres chevaux fossiles.
Il apparaît ainsi qu’alors que la diversité génétique était restée stable pendant 4 millénaires, celle-ci a baissé de 16% au cours des 200 à 400 dernières années. Cette période coïnciderait avec de forts changements de pratiques d’élevage marqués par une réduction du nombre de chevaux reproducteurs, entraînant une réduction de la taille efficace de la population, c’est-à-dire le nombre d’individus d’une population idéale chez laquelle on observerait un degré de dérive génétique équivalent à celui de la population réelle. Cette réduction n’est pas sans impact : la théorie prédit que les petites populations seraient en effet marquées par une atténuation de la sélection purifiante (sélection contre le maintien des allèles délétères), entraînant l’accumulation d’un fardeau génétique. La comparaison des patrons de sélection sur les sites synonymes et sur les sites non synonymes ainsi que sur ceux classés comme délétères par comparaison avec les variations conservées chez les espèces de Vertébrés a permis de vérifier cet attendu théorique chez les populations de chevaux : le fardeau génétique a bien augmenté chez les chevaux modernes, corrélativement à la perte de diversité. Cette réduction de diversité s’expliquerait par des stratégies drastiques de sélection d’étalons pour la reproduction. La diversité nucléotidique sur le chromosome Y, transmis par les étalons, diminue ainsi, à la fois en Asie et en Europe, au cours des deux derniers millénaires et chute aux niveaux actuels à partir de 850-1 350 de l’ère commune (anciennement appelée période après Jésus-Christ).
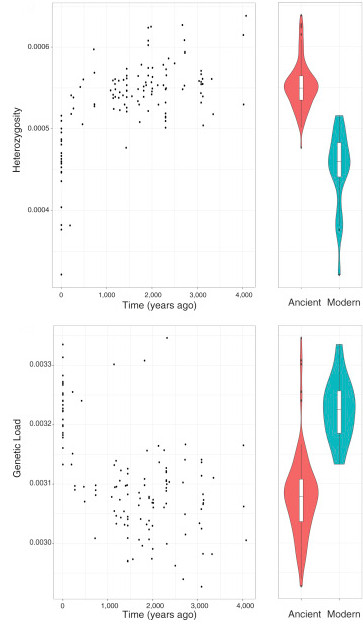
Évolution de la diversité et du fardeau génétique chez le cheval domestique au cours du temps. En haut, la diversité, évaluée par l’hétérozygotie, chute brusquement chez les chevaux modernes par comparaison aux chevaux anciens. En bas, le fardeau génétique augmente corrélativement à la baisse de diversité, chez les chevaux modernes.
L’étude des relations phylogénétiques entre chevaux anciens et modernes est très informative quant aux échanges survenus les siècles passés. En plus des chevaux domestiques et des chevaux de Przewalski, les échantillons les plus anciens indiquent l’existence de deux autres lignées, aujourd’hui éteintes, de chevaux sauvages, l’une dans la péninsule ibérique, l’autre en Sibérie. Bien que présentes à l’époque de la domestication du cheval domestique, ces deux lignées n’auraient eu qu’une contribution marginale à la diversité des chevaux domestiques modernes, permettant de rejeter l’hypothèse d’un centre de domestication du cheval dans la péninsule ibérique.
En se focalisant sur la phylogénie des chevaux domestiques, on remarque que les poneys Shetlands et les chevaux Islandais modernes se classent à proximité de chevaux anciens du Nord de l’Europe. Ces deux races de chevaux prendraient peut-être leur origine dans les conquêtes vikings des VIIIe-XIe siècles. Le clade formé par ces chevaux est un clade frère de chevaux anciens européens, de la période Gallo-Romaine ou de la Tène (culture archéologique du 2nd Age du fer), traduisant une certaine cohésion génétique des chevaux européens anciens. Les chevaux modernes européens, autres que les poneys Shetlands et les chevaux Islandais, se retrouvent dans un autre clade, qui apparaîtrait en Europe au IXe siècle en Croatie, à une époque où ce fond génétique est encore absent en Europe du Nord. Sachant que cette période correspond à de fréquents raids arabes sur les côtes méditerranéennes et que ce clade correspond aussi à celui de chevaux persans sassanides des IVe et Ve siècles, ces résultats suggèrent une forte influence génétique des chevaux persans en Europe à partir du IXe siècle. Des résultats similaires ont été relevés en Asie avec le remplacement des fonds génétiques pré-existants en Asie centrale et en Mongolie par les chevaux d’origine persane à partir des VIIIe-IXe siècles.
Cet échantillon est aussi une opportunité de comprendre les gènes sélectionnés et par conséquent les caractères qui auraient été recherchés au cours de l’histoire du cheval domestique. Ainsi, la comparaison des fréquences alléliques entre les chevaux anciens asiatiques et européens et les chevaux byzantins de l’époque post-VIIe-IXe siècles, déjà largement marqués par l’introgression des chevaux d’origine persane, montre que les gènes impliqués dans la morpho-anatomie auraient beaucoup évolué sous l’influence des chevaux d’origine persane. Le gène MSTN impliqué dans la vitesse serait aussi un candidat sélectionné chez ces chevaux byzantins d’origine persane. Plus récemment, au cours du dernier millénaire, la sélection d’allèle à ce gène MSTN, mais aussi à deux autres gènes PDK4 et ACN9 connus pour influencer la vitesse des chevaux, confirme que l’accroissement de la vitesse de ses montures a été une préoccupation majeure de l’Homme.
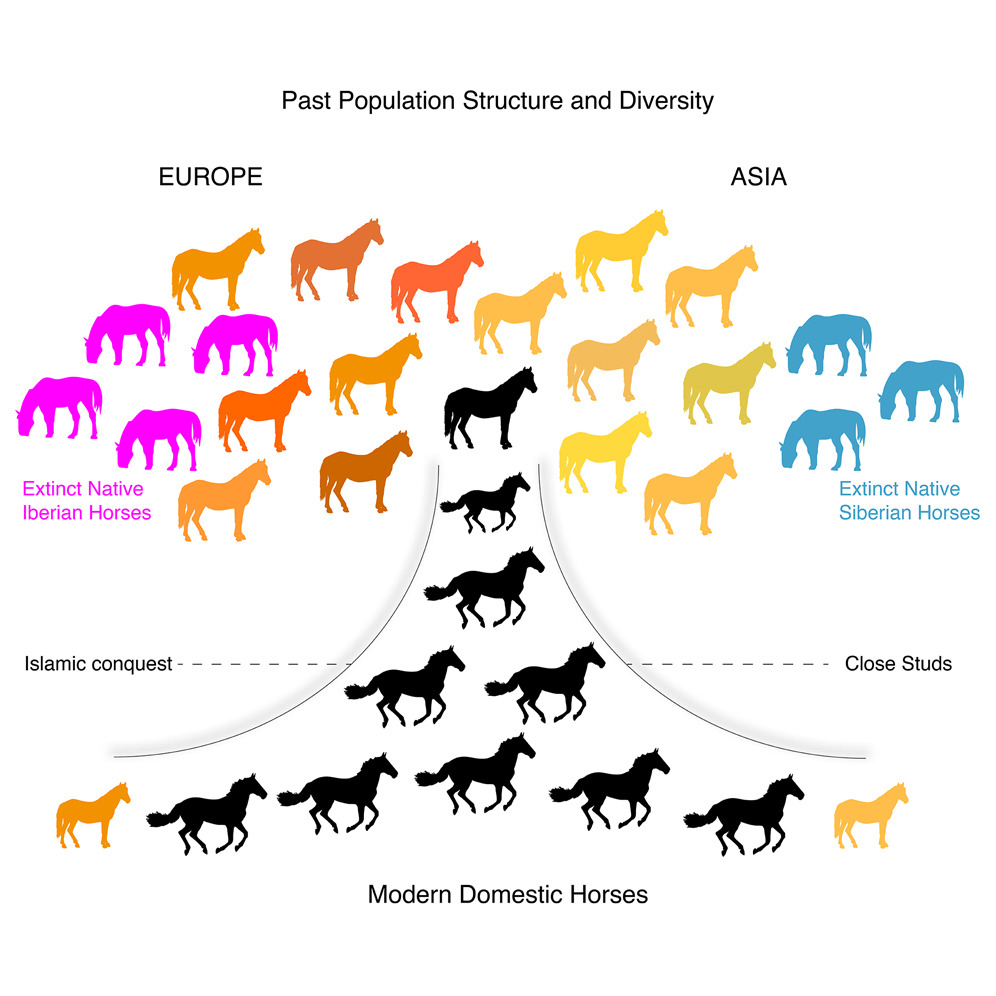
Synthèse de l’histoire démographique du cheval cultivé, publiée dans l’article. Les conquêtes islamiques auraient entraîné une diffusion des chevaux de types persans qui auraient remplacé presque toutes les lignées de chevaux anciens présents en Asie et en Europe.
Références de l’article :