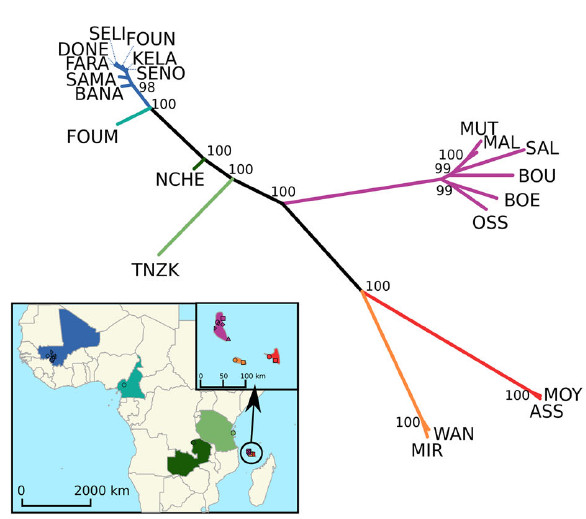
L’archéologie montre que quand les peuples de langue papoue et les Aborigènes d’Australie réalisent le premier peuplement humain de l’Océanie il y a environ 50 000 ans, ils ne colonisent que l’Océanie proche (jusqu’aux Iles Salomon). Une seconde vague de migration, composée de peuples de langues austronésiennes, survient beaucoup plus récemment il y a 5 000 ans et se poursuit cette fois-ci jusqu’à l’Océanie lointaine, notamment le Vanuatu, la Nouvelle-Calédonie, Fidji, Tonga et Samoa. Ainsi, les restes archéologiques font remonter les premiers peuplements aux îles Samoa il y a seulement 2 750 à 2 880 ans avant une histoire démographique assez floue jusqu’à l’arrivée des premiers Européens au XVIIIe siècle.

Trois jeunes femmes samoanes de 1900. Photographie de Ernst von Hesse-Wartegg (1854-1918), publiée dans le livre Samoa, Bismarckarchipel und Neuguinea – Drei deutsche Kolonien in der Südsee; (Allemagne, 1902). Photographie du domaine public.
L’étude de Daniel N. Harris et ses collaborateurs américains, néo-zélandais et samoans, publiée dans le journal Proceedings of the National Academy of Sciences of the United States of America vise à comprendre l’évolution démographique des Samoans au cours de leur Histoire. Un total de 1 197 génomes complets de Samoans ont été séquencés et soumis à des analyses de génomique des populations.
L’analyse génétique confirme que les Samoans sont très majoritairement d’origine austronésienne, avec de rares introgressions de populations d’Europe de l’Ouest, du Sud de l’Asie et d’Afrique de l’Ouest. Toutefois, les Samoans auraient en moyenne 24 % d’origine papoue. Cela suggère un phénomène déjà montré pour d’autres peuples océaniens : avant d’arriver en Océanie lointaine, les peuples de langues austronésiennes se seraient hybridés avec les peuples papous rencontrés en Océanie proche. L’étude montre aussi que l’origine papoue est corrélée à l’origine dénisovienne. Les Dénisoviens ou Hommes du Denisova représentent une espèce d’Hommes “fossiles” apparentés aux Hommes de Néanderthal. Les Papous sont issus de populations humaines s’étant hybridées avec ces Dénisoviens, peut-être en Asie du Sud-Est, et conservent 3 à 6% de leur génome hérité de leurs ancêtres dénisoviens. Par leur ascendance papoue, les Samoans auraient aussi hérité de quelques portions de génome de Dénisoviens, à un degré moindre.
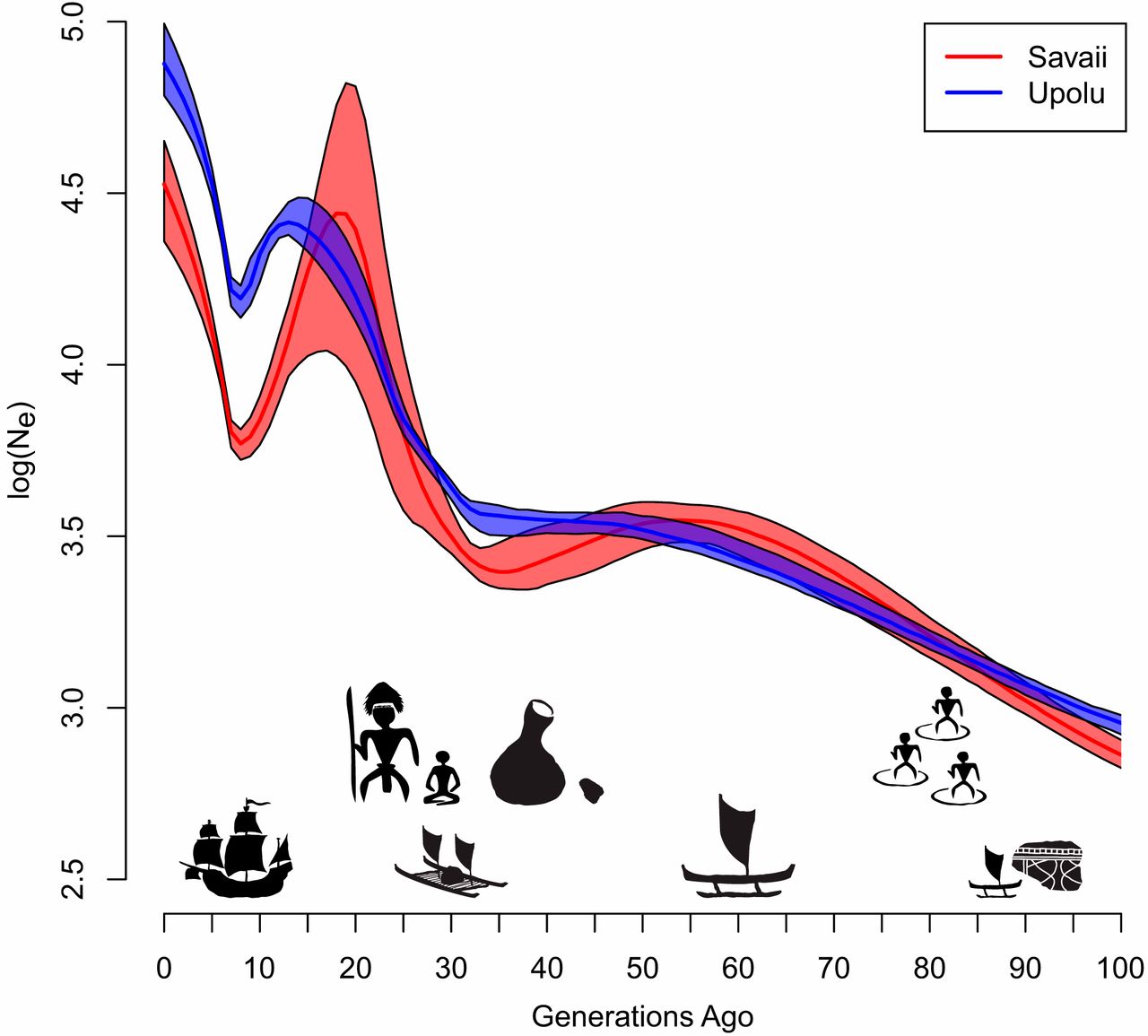
Variation de l’effectif efficace Ne, représenté ici après une transformation logarithmique, dans la populations des deux îles de Savai’i (rouge) et d’Upolu (bleu) en fonction du nombre de générations depuis le présent. La variation de l’effectif efficace est indiquée par une ligne. Cette ligne est comprise dans un polygone qui symbolise son intervalle de confiance à 95%. Les principaux événements historiques supposés sur les îles Samoa sont indiqués par des pictogrammes. Il y a 92 à 96 générations (2 750 à 2 880 ans), l’île d’Upolu est colonisée par des peuples de culture Lapita. Il y a 70 à 90 générations (2 100 à 2 700 ans), les autres îles sont colonisées par de petits groupes isolés. Il y a 50 à 67 générations (1 500 à 2 000 ans), possibles migrations d’individus depuis les îles Carolines (Micronésie). Il y a 33 à 50 générations (1 000 à 1 500 ans), les poteries disparaissent, en même temps qu’un premier goulot d’étranglement majeur sur les îles Samoa. Il y a 27 à 33 générations (800 à 1 000 ans), les voyages entre archipels du Pacifique s’intensifient. Il y a 17 à 27 générations (500 à 800 ans), des indices montrent une forte activité humaine aux Samoa : développement de chefferies, de monuments, d’infrastructures agricoles en terrasses, etc. Il y a 8 générations (230 ans), arrivée des premiers Européens qui apportent des maladies sur les îles, aboutissant au 2e goulot d’étranglement majeur sur les îles Samoa.
Le modèle démographique construit à partir des données génétiques donne des indications sur les changements d’effectif efficace de la population des Samoa. Cet effectif efficace représente l’effectif d’une population théorique qui serait soumise à la même dérive génétique que la population réelle. Les variations de l’effectif efficace sont à mettre en relation avec les événements historiques connus. L’histoire démographique des Samoa commencerait par une période de croissance débutée il y a 100 générations. En considérant un temps intergénérationnel de 30 ans, cela représente environ 3 000 ans. Ce temps est cohérent avec les premiers restes archéologiques et l’apparition de la culture Lapita, connue pour ses poteries décorées. Pendant 70 générations, l’effectif efficace des Samoans serait resté très faible (entre 700 et 3 440 individus), ce qui suggère de petites populations sur les îles Samoa. Cela confirme aussi la faible quantité de sites archéologiques découverts sur les îles Samoa en comparaison des îles Fidji et Tonga qui ont connu une croissance démographique plus précoce.
Alors que l’histoire démographique était jusqu’alors restée similaire entre les deux îles, l’histoire démographique de l’île d’Upolu, qui compte aujourd’hui les villes les plus importantes des Samoa, diverge de celle de Savai’i à partir de 30 à 35 générations avant aujourd’hui, soit il y a 900 à 1 050 ans, après une longue période de goulot d’étranglement (réduction de la taille de la population). Après cette date, la population des Samoa est marquée par une forte période de croissance exponentielle. Ce brusque changement d’effectif efficace suggère un changement démographique important il y a 900 à 1 050 ans. La période du goulot d’étranglement commence après l’arrivée supposée de peuples venus des îles Carolines et/ou de Micronésie. Que s’est-il passé sur cette période ? Y a-t-il eu un remplacement de la population initiale des Samoa par ces nouveaux arrivants ? Les deux populations se sont-elles mélangées ? On manque pour le moment de données susceptibles d’aider à répondre à ces questions, notamment d’ADN ancien qui pourrait être extrait des squelettes des habitants de l’époque.
Beaucoup plus récemment, à la fin de la période de croissance exponentielle, on est capable d’identifier un deuxième goulot d’étranglement il y a environ 10 générations, c’est-à-dire 300 ans. Cela correspond à l’époque des premiers contacts avec les Européens qui ont apporté des maladies auxquelles le système immunitaire des Samoans n’était pas préparé, notamment la rougeole qui se révèle dangereuse lorsque cette maladie virale est contractée à l’âge adulte.
Références de l’article